
本文为Yijun Yang, Zhao-Yang Wang的《Medical World Model: Generative Simulation of Tumor Evolution for Treatment Planning》一文的相关复现。
论文概要
本文通过以肿瘤图像数据作为基础,构建了基于肿瘤发展的时序世界模型。该模型是第一种能够基于临床决策来预测未来疾病状态的医学世界模型。
MeWM由三个主要组件构成:
政策模型,该模型基于视觉语言架构,能够根据患者的当前状态及具体的临床情境生成可能的操作方案;
动态模型,该模型能够模拟肿瘤的发展过程,通过生成式建模来预测在不同治疗条件下肿瘤如何进展或衰退;
逆向动态模型,该模型能够对模拟后的肿瘤进行生存风险分析,并定量评估治疗的疗效。
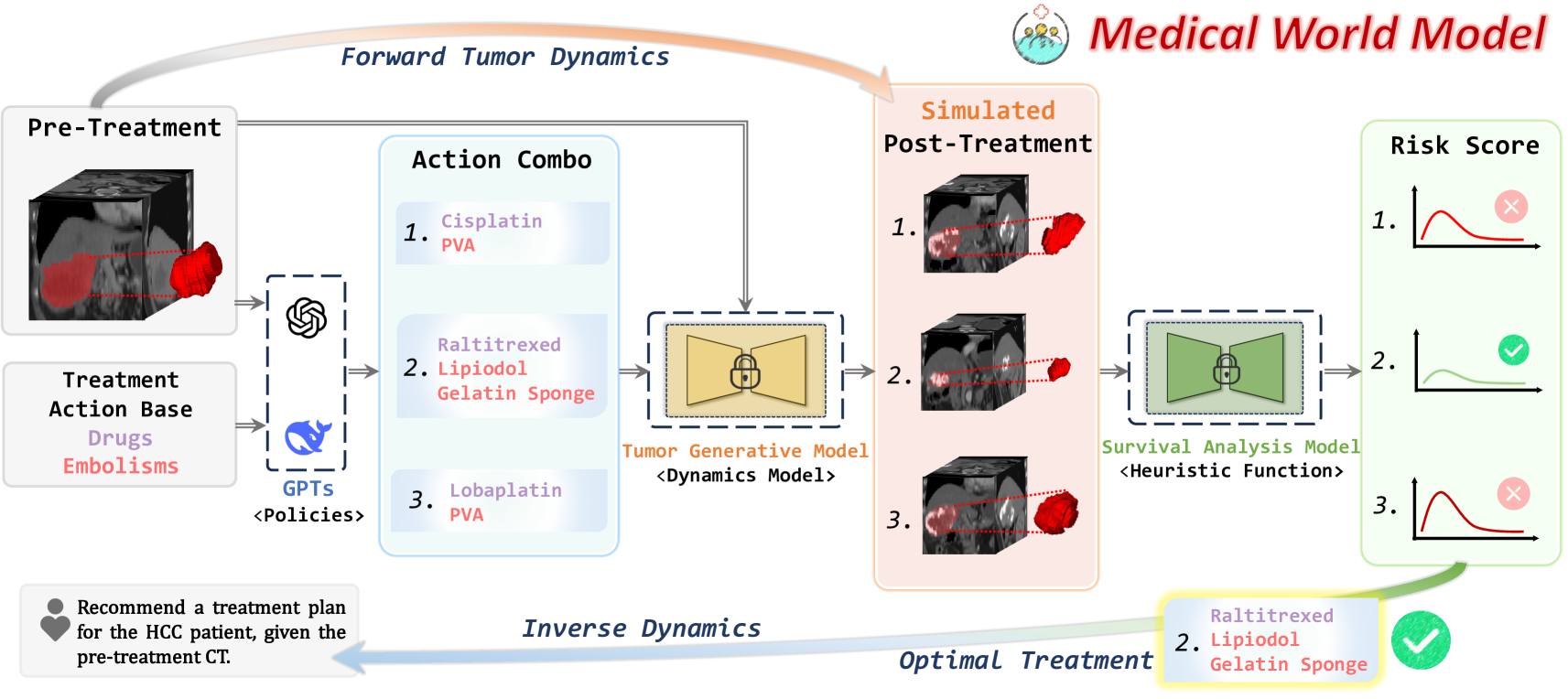
除了前向模拟之外,该系统还借助分割模型来启发式地寻找最优方案。通过整合这些组件,MeWM提供了一种全面的决策框架:它能够模拟出真实世界的肿瘤发展情况。在放射科医生测试中,该模型的表现优于那些类似GPT的专用模型。在经动脉化疗栓塞术方案探索任务上,该模型的性能提升超过了10%,在F1分数上表现尤为突出。
复现
项目开源地址


复现步骤
环境配置
首先克隆:
BASH
git clone https://github.com/scott-yjyang/MeWM.git
然后创建环境,安装依赖:
BASH
cd MeWM
conda create -n mewm_py311 python=3.11
conda activate mewm_py311
conda install pip
pip install -r requirements.txt
有报错的话,以下几个包手动安装一下:
PLAINTEXT
conda install conda-forge::pyairports
pip install git+https://ghproxy.net/https://github.com/Radiomics/pyradiomics.git@v3.1.0
python -m pip install "setuptools<82.0.0" --force-reinstall
然后配置下HF的镜像:
BASH
export HF_ENDPOINT=https://hf-mirror.com
然后下载解码模型:
BASH
mkdir Synthesis/Diffusion/pretrained_models
cd Synthesis/Diffusion/pretrained_models
pip install -U "huggingface-hub>=0.23.2,<1.0" hf_transfer
hf auth login
hf download MrGiovanni/DiffTumor AutoencoderModel/AutoencoderModel.ckpt --local-dir ./
mv AutoencoderModel.ckpt ../
预处理
首先下载HCC-TACE-Seg数据集(在此处公开,需要用TCIA Data Retriever下载,大约30G),然后用以下代码进行预处理:
PYTHON
#!/usr/bin/env python3
"""
HCC-TACE DICOM → NIfTI 预处理脚本
将 TCIA 下载的 DICOM 数据转换为训练代码期望的格式
用法:
python preprocess_hcc_tace.py --dicom_root /path/to/hcc_tace_seg --output_root /path/to/output
输出目录结构:
output/
├─ HCC_tace_response_trans_pre/ # 治疗前 CT
│ ├─ HCC_001_ap.nii.gz
│ ├─ HCC_001_pvp.nii.gz
│ └─ ...
├─ HCC_tace_response_trans_pre_mask/ # 治疗前肿瘤 mask
├─ HCC_tace_response_trans_post/ # 治疗后 CT
├─ HCC_tace_response_trans_post_mask/# 治疗后肿瘤 mask
├─ train_paired_all_hcc_under_50_new.txt # 训练列表
└─ val_paired_all_hcc_under_50_new.txt # 验证列表
"""
import os
import sys
import argparse
import subprocess
import shutil
import glob
import numpy as np
import pydicom
from pydicom.errors import InvalidDicomError
from collections import defaultdict
from concurrent.futures import ThreadPoolExecutor, as_completed
from tqdm import tqdm
import random
# ============================================================
# 1. DICOM 扫描与识别
# ============================================================
def scan_dicom_structure(dicom_root):
"""
扫描 DICOM 目录结构,返回每个患者的 study/series 信息
Returns:
patients: dict {
patient_id: {
'studies': {
study_uid: {
'series': {
series_uid: {
'description': str,
'modality': str,
'num_files': int,
'first_file': str,
'sample_dicom': pydicom.Dataset
}
}
}
}
}
}
"""
patients = {}
for patient_dir in sorted(os.listdir(dicom_root)):
patient_path = os.path.join(dicom_root, patient_dir)
if not os.path.isdir(patient_path) or not patient_dir.startswith('HCC_'):
continue
patient_id = patient_dir
patients[patient_id] = {'studies': {}}
# 遍历 study 目录(第一层数字目录)
for study_dir in sorted(os.listdir(patient_path)):
study_path = os.path.join(patient_path, study_dir)
if not os.path.isdir(study_path):
continue
study_uid = study_dir
# 确保 study 条目存在
if study_uid not in patients[patient_id]['studies']:
patients[patient_id]['studies'][study_uid] = {'series': {}}
# 遍历 series 目录(第二层数字目录)
for series_dir in sorted(os.listdir(study_path)):
series_path = os.path.join(study_path, series_dir)
if not os.path.isdir(series_path):
continue
series_uid = series_dir
# 查找该 series 的第一个 DICOM 文件
dcm_files = []
for f in sorted(os.listdir(series_path)):
if f.endswith('.dcm') or not os.path.splitext(f)[1]:
dcm_files.append(os.path.join(series_path, f))
if not dcm_files:
continue
# 读取第一个 DICOM 获取元数据
try:
ds = pydicom.dcmread(dcm_files[0], stop_before_pixels=True)
except Exception:
continue
series_desc = getattr(ds, 'SeriesDescription', 'NO_DESC').strip()
modality = getattr(ds, 'Modality', 'UNKNOWN')
patients[patient_id]['studies'][study_uid]['series'][series_uid] = {
'description': series_desc,
'modality': modality,
'num_files': len(dcm_files),
'first_file': dcm_files[0],
'series_path': series_path,
}
return patients
def print_patient_info(patients, max_patients=5):
"""打印前几个患者的信息用于调试"""
count = 0
for pid in sorted(patients.keys()):
if count >= max_patients:
break
count += 1
print(f"\n{'='*60}")
print(f"患者: {pid}")
print(f" Study 数量: {len(patients[pid]['studies'])}")
for suid, study in patients[pid]['studies'].items():
print(f" Study: {suid}")
for seuid, series in study['series'].items():
desc = series['description']
mod = series['modality']
n = series['num_files']
print(f" Series: {seuid} | {mod} | {desc} ({n} files)")
# ============================================================
# 2. DICOM → NIfTI 转换
# ============================================================
def check_dcm2niix():
"""检查 dcm2niix 是否可用"""
return shutil.which('dcm2niix') is not None
def convert_series_to_nifti(series_path, output_dir, output_name):
"""
使用 dcm2niix 将单个 DICOM series 转为 NIfTI
"""
os.makedirs(output_dir, exist_ok=True)
# 临时输出
tmp_dir = os.path.join(output_dir, '.tmp_dcm2niix')
os.makedirs(tmp_dir, exist_ok=True)
cmd = [
'dcm2niix',
'-z', 'y', # 压缩为 .nii.gz
'-f', output_name, # 输出文件名
'-o', tmp_dir, # 输出目录
'-b', 'n', # 不创建 BIDS 格式
'-m', 'n', # 不合并 2D slices
series_path
]
try:
result = subprocess.run(cmd, capture_output=True, text=True, timeout=120)
# 查找生成的 nii.gz 文件
nii_files = glob.glob(os.path.join(tmp_dir, f'{output_name}*.nii.gz'))
if nii_files:
# 移动到目标目录
dest = os.path.join(output_dir, f'{output_name}.nii.gz')
shutil.move(nii_files[0], dest)
# 清理
shutil.rmtree(tmp_dir, ignore_errors=True)
return dest
else:
# 可能 dcm2niix 添加了后缀
all_nii = glob.glob(os.path.join(tmp_dir, '*.nii.gz'))
if all_nii:
dest = os.path.join(output_dir, f'{output_name}.nii.gz')
shutil.move(all_nii[0], dest)
shutil.rmtree(tmp_dir, ignore_errors=True)
return dest
print(f" [WARN] dcm2niix 未生成文件: {series_path}")
shutil.rmtree(tmp_dir, ignore_errors=True)
return None
except subprocess.TimeoutExpired:
print(f" [ERROR] dcm2niix 超时: {series_path}")
shutil.rmtree(tmp_dir, ignore_errors=True)
return None
except Exception as e:
print(f" [ERROR] dcm2niix 异常: {e}")
shutil.rmtree(tmp_dir, ignore_errors=True)
return None
def convert_with_pydicom_simple(series_path, output_path):
"""
备选方案: 使用 pydicom + nibabel 进行简单转换(无需 dcm2niix)
"""
try:
import nibabel as nib
dcm_files = []
for f in sorted(os.listdir(series_path)):
full_path = os.path.join(series_path, f)
try:
pydicom.dcmread(full_path, stop_before_pixels=True)
dcm_files.append(full_path)
except Exception:
continue
if not dcm_files:
return None
# 按 InstanceNumber 或 SliceLocation 排序
slices = []
for f in dcm_files:
try:
ds = pydicom.dcmread(f)
slices.append(ds)
except Exception:
continue
if not slices:
return None
# 尝试按 SliceLocation 排序
try:
slices.sort(key=lambda x: float(x.SliceLocation))
except Exception:
try:
slices.sort(key=lambda x: int(x.InstanceNumber))
except Exception:
pass
# 构建 3D volume
pixel_arrays = []
for s in slices:
pixel_arrays.append(s.pixel_array)
volume = np.stack(pixel_arrays, axis=0).astype(np.float32)
# 构建 affine(简化版)
try:
first_slice = slices[0]
spacing = [float(s) for s in first_slice.PixelSpacing]
spacing.append(float(first_slice.SliceThickness) if hasattr(first_slice, 'SliceThickness') else 1.0)
except Exception:
spacing = [1.0, 1.0, 1.0]
affine = np.diag(spacing + [1.0])
# 保存
img = nib.Nifti1Image(volume, affine)
nib.save(img, output_path)
return output_path
except ImportError:
print(" [ERROR] 需要安装 nibabel: pip install nibabel")
return None
except Exception as e:
print(f" [ERROR] pydicom 转换失败: {e}")
return None
# ============================================================
# 3. 系列类型识别
# ============================================================
def classify_series(description, modality):
"""
根据 SeriesDescription 和 Modality 分类系列类型
Returns:
'arterial_ct', 'pvp_ct', 'pre_ct' (non-contrast, skip),
'tumor_mask', 'other_ct', 'other'
"""
desc_lower = description.lower()
# ============================================================
# 肿瘤 mask 识别
# ============================================================
# SEG/RTSTRUCT 模态直接识别为 mask
if modality.upper() in ('SEG', 'RTSTRUCT', 'RTSEG'):
return 'tumor_mask'
# 描述中包含 mask/segmentation 关键词(部分数据可能以 CT 存储 mask)
if any(kw in desc_lower for kw in ['segmentation', 'tumor mask', 'lesion mask']):
return 'tumor_mask'
# 非 CT 模态 → 跳过
if modality.upper() != 'CT':
return 'other'
# ============================================================
# 排除平扫(非对比剂)
# ============================================================
if 'pre liver' in desc_lower:
return 'pre_ct'
# ============================================================
# 动脉期识别
# ============================================================
# (AP), AP, Arterial, C-A-P (Contrast-Arterial-Phase)
arterial_patterns = [
'(ap)', # LIVER 3 PHASE (AP)
'arterial', # Arterial Phase
'c-a-p', # C-A-P = Contrast Arterial Phase
' ap ', # standalone AP
]
if any(p in desc_lower for p in arterial_patterns):
return 'arterial_ct'
# 描述以 "ap" 开头或以 "ap)" 结尾
if desc_lower.startswith('ap ') or desc_lower.endswith('(ap') or desc_lower == 'ap':
return 'arterial_ct'
# ============================================================
# 门脉期识别
# ============================================================
pvp_patterns = [
'(pvp)', # LIVER 3 PHASE (PVP)
'portal venous',
'portal vein',
' pvp ',
]
if any(p in desc_lower for p in pvp_patterns):
return 'pvp_ct'
# ============================================================
# 延迟期 → 跳过
# ============================================================
if any(kw in desc_lower for kw in ['delay', 'delayed', 'equilibrium']):
return 'other'
# ============================================================
# 多期混合扫描(无明确分期)→ 保留作为备选
# 如 "3 PHASE LIVER (ABD)" 或 "3 PHASE LIVER A/P"
# ============================================================
if any(kw in desc_lower for kw in ['3 phase', 'multi phase', 'triphasic', 'a/p']):
return 'other_ct'
# ============================================================
# Recon N: 前缀 → 去掉前缀后重新判断
# ============================================================
for prefix in ['recon 2: ', 'recon 3: ', 'recon 4: ', 'recon 1: ']:
if desc_lower.startswith(prefix):
base_desc = desc_lower[len(prefix):].strip()
if base_desc:
return classify_series(base_desc, modality)
# ============================================================
# 完全无法识别 → 保留作为备选
# ============================================================
return 'other_ct'
def identify_study_type(study_uid, series_dict, all_studies):
"""
判断 study 是治疗前还是治疗后
通常 HCC_XXX 的第一个 study 是治疗前,第二个是治疗后
"""
study_indices = list(all_studies.keys())
try:
idx = study_indices.index(study_uid)
# 第一个 study → pre,第二个 → post
return 'pre' if idx == 0 else 'post'
except ValueError:
return 'unknown'
# ============================================================
# 4. 主处理流程
# ============================================================
def process_dataset(dicom_root, output_root, use_simple_converter=False):
"""
主处理流程
"""
print("=" * 60)
print("Step 1: 扫描 DICOM 目录结构...")
print("=" * 60)
patients = scan_dicom_structure(dicom_root)
print(f"\n发现 {len(patients)} 个患者")
print_patient_info(patients, max_patients=3)
# ============================================================
# 创建输出目录
# ============================================================
pre_ct_dir = os.path.join(output_root, 'HCC_tace_response_trans_pre')
pre_mask_dir = os.path.join(output_root, 'HCC_tace_response_trans_pre_mask')
post_ct_dir = os.path.join(output_root, 'HCC_tace_response_trans_post')
post_mask_dir = os.path.join(output_root, 'HCC_tace_response_trans_post_mask')
for d in [pre_ct_dir, pre_mask_dir, post_ct_dir, post_mask_dir]:
os.makedirs(d, exist_ok=True)
# ============================================================
# Step 2: DICOM → NIfTI 转换
# ============================================================
print("\n" + "=" * 60)
print("Step 2: DICOM → NIfTI 转换...")
print("=" * 60)
if not use_simple_converter and not check_dcm2niix():
print("[WARN] dcm2niix 未安装,回退到 pydicom 转换")
print(" 建议安装: conda install -c conda-forge dcm2niix")
use_simple_converter = True
data_entries = [] # 存储每对数据的信息
for patient_id in sorted(patients.keys()):
print(f"\n处理患者: {patient_id}")
patient_info = patients[patient_id]
studies = patient_info['studies']
# 按顺序确定 pre/post study
study_uids = sorted(studies.keys())
pre_study_uid = study_uids[0] if len(study_uids) > 0 else None
post_study_uid = study_uids[1] if len(study_uids) > 1 else None
# ============================================================
# 收集所有 study 中的 CT 和 mask
# ============================================================
all_ct = {} # {study_uid: {'ap': name, 'pvp': name, 'other': name}}
all_masks = {} # {study_uid: name}
for study_uid in study_uids:
ct_files = {}
mask_file = None
for series_uid, series_info in studies[study_uid]['series'].items():
series_type = classify_series(series_info['description'], series_info['modality'])
series_path = series_info['series_path']
# --- CT 系列 ---
if series_type in ('arterial_ct', 'pvp_ct', 'other_ct'):
if series_type == 'arterial_ct':
suffix = '_artery'
elif series_type == 'pvp_ct':
suffix = '_pvp'
else:
suffix = '_ct'
out_name = f'{patient_id}{suffix}'
if use_simple_converter:
out_ct_dir = pre_ct_dir if study_uid == pre_study_uid else post_ct_dir
out_path = os.path.join(out_ct_dir, f'{out_name}.nii.gz')
result = convert_with_pydicom_simple(series_path, out_path)
else:
out_ct_dir = pre_ct_dir if study_uid == pre_study_uid else post_ct_dir
result = convert_series_to_nifti(series_path, out_ct_dir, out_name)
if result:
key = 'ap' if series_type == 'arterial_ct' else ('pvp' if series_type == 'pvp_ct' else 'other')
ct_files[key] = out_name + '.nii.gz'
print(f" ✓ CT ({series_type}): {out_name}")
# --- Mask 系列 ---
elif series_type == 'tumor_mask':
out_name = f'{patient_id}_tumor'
out_mask_dir = pre_mask_dir if study_uid == pre_study_uid else post_mask_dir
if use_simple_converter:
out_path = os.path.join(out_mask_dir, f'{out_name}.nii.gz')
result = convert_with_pydicom_simple(series_path, out_path)
else:
result = convert_series_to_nifti(series_path, out_mask_dir, out_name)
if result:
mask_file = out_name + '.nii.gz'
study_label = 'PRE' if study_uid == pre_study_uid else 'POST'
print(f" ✓ Mask ({study_label}): {out_name}")
if ct_files:
all_ct[study_uid] = ct_files
if mask_file:
all_masks[study_uid] = mask_file
# ============================================================
# Mask 共享:如果只有一个 study 有 mask,复制到另一个 study
# ============================================================
if len(all_masks) == 1:
only_uid = list(all_masks.keys())[0]
other_uid = pre_study_uid if only_uid == post_study_uid else post_study_uid
if other_uid:
src_mask_name = all_masks[only_uid]
src_dir = pre_mask_dir if only_uid == pre_study_uid else post_mask_dir
dst_dir = post_mask_dir if only_uid == pre_study_uid else pre_mask_dir
src_path = os.path.join(src_dir, src_mask_name)
dst_path = os.path.join(dst_dir, src_mask_name)
if os.path.exists(src_path) and not os.path.exists(dst_path):
shutil.copy2(src_path, dst_path)
all_masks[other_uid] = src_mask_name
print(f" → Mask 共享: {src_mask_name} (PRE ↔ POST)")
# ============================================================
# 选择最佳 CT 配对
# 优先级: arterial > pvp > other_ct
# ============================================================
def pick_best_ct(ct_dict):
return ct_dict.get('ap') or ct_dict.get('pvp') or ct_dict.get('other')
pre_ct_sel = pick_best_ct(all_ct.get(pre_study_uid, {})) if pre_study_uid else None
post_ct_sel = pick_best_ct(all_ct.get(post_study_uid, {})) if post_study_uid else None
pre_mask_sel = all_masks.get(pre_study_uid) if pre_study_uid else None
post_mask_sel = all_masks.get(post_study_uid) if post_study_uid else None
if pre_ct_sel and post_ct_sel and pre_mask_sel and post_mask_sel:
data_entries.append({
'patient_id': patient_id,
'pre_ct': pre_ct_sel,
'pre_mask': pre_mask_sel,
'post_ct': post_ct_sel,
'post_mask': post_mask_sel,
})
print(f" → 配对成功: pre={pre_ct_sel}/{pre_mask_sel}, post={post_ct_sel}/{post_mask_sel}")
else:
missing = []
if not pre_ct_sel: missing.append('pre_ct')
if not post_ct_sel: missing.append('post_ct')
if not pre_mask_sel: missing.append('pre_mask')
if not post_mask_sel: missing.append('post_mask')
print(f" [SKIP] 缺失: {missing}")
# ============================================================
# Step 3: 创建数据列表文件
# ============================================================
print("\n" + "=" * 60)
print("Step 3: 创建数据列表文件...")
print("=" * 60)
print(f"成功配对: {len(data_entries)} 个患者")
if not data_entries:
print("\n[ERROR] 没有成功配对的数据!")
print("请检查:")
print(" 1. DICOM SeriesDescription 是否正确识别")
print(" 2. 每个患者是否都有 pre/post 的 CT 和 mask")
print("\n运行 --inspect 模式查看详细 DICOM 信息:")
print(" python preprocess_hcc_tace.py --inspect --dicom_root <path>")
return
# 划分训练集/验证集 (90/10)
random.seed(42)
random.shuffle(data_entries)
split_idx = int(len(data_entries) * 0.9)
train_entries = data_entries[:split_idx]
val_entries = data_entries[split_idx:]
# 生成数据列表格式:
# {pre_ct}\t{pre_mask}\t\t\t{post_ct}\t{post_mask}\t\t\t\t{treatment_text}
# 列索引: 0 1 2 3 4 5 6 7 8 9
def write_data_list(entries, output_path):
with open(output_path, 'w') as f:
for entry in entries:
# 构建相对路径
pre_ct_rel = f'HCC_tace_response_trans_pre/{entry["pre_ct"]}'
pre_mask_rel = f'HCC_tace_response_trans_pre_mask/{entry["pre_mask"]}'
post_ct_rel = f'HCC_tace_response_trans_post/{entry["post_ct"]}'
post_mask_rel = f'HCC_tace_response_trans_post_mask/{entry["post_mask"]}'
# 文本描述(使用占位符,后续可根据实际临床数据填充)
treatment_text = 'TACE treatment'
# Tab 分隔的格式:共 10 列
line = '\t'.join([
pre_ct_rel, # 0: pre image
pre_mask_rel, # 1: pre label
'', # 2: (unused)
'', # 3: (unused)
post_ct_rel, # 4: post image
post_mask_rel, # 5: post label
'', # 6: (unused)
'', # 7: (unused)
'', # 8: (unused)
treatment_text, # 9: text description
])
f.write(line + '\n')
print(f" 写入 {len(entries)} 条记录 → {output_path}")
train_list_path = os.path.join(output_root, 'train_paired_all_hcc_under_50_new.txt')
val_list_path = os.path.join(output_root, 'val_paired_all_hcc_under_50_new.txt')
write_data_list(train_entries, train_list_path)
write_data_list(val_entries, val_list_path)
# ============================================================
# Step 4: 输出总结
# ============================================================
print("\n" + "=" * 60)
print("预处理完成!")
print("=" * 60)
print(f"""
输出目录: {output_root}
├─ HCC_tace_response_trans_pre/ ({len(set(e['pre_ct'] for e in data_entries))} 文件)
├─ HCC_tace_response_trans_pre_mask/ ({len(set(e['pre_mask'] for e in data_entries))} 文件)
├─ HCC_tace_response_trans_post/ ({len(set(e['post_ct'] for e in data_entries))} 文件)
├─ HCC_tace_response_trans_post_mask/ ({len(set(e['post_mask'] for e in data_entries))} 文件)
├─ train_paired_all_hcc_under_50_new.txt ({len(train_entries)} 条)
└─ val_paired_all_hcc_under_50_new.txt ({len(val_entries)} 条)
下一步:
1. 将输出目录 SCP/RSYNC 到服务器上的训练数据目录
2. 创建 cross_eval/liver/ 目录并放入数据列表文件
3. 确保 clinical data CSV 文件存在
4. 运行训练命令
""")
# ============================================================
# 5. 检查模式:查看 DICOM 元数据
# ============================================================
def inspect_mode(dicom_root, max_patients=10):
"""检查模式:详细打印 DICOM 元数据以便调试"""
patients = scan_dicom_structure(dicom_root)
count = 0
for pid in sorted(patients.keys()):
if count >= max_patients:
break
count += 1
print(f"\n{'='*60}")
print(f"患者: {pid}")
for suid, study in patients[pid]['studies'].items():
study_idx = list(patients[pid]['studies'].keys()).index(suid)
study_type = 'PRE' if study_idx == 0 else 'POST' if study_idx == 1 else 'UNKNOWN'
print(f" [{study_type}] Study: {suid}")
for seuid, series in study['series'].items():
desc = series['description']
mod = series['modality']
n = series['num_files']
series_type = classify_series(desc, mod)
print(f" Series: {seuid} | {mod:6s} | {series_type:12s} | {desc} ({n} files)")
# ============================================================
# Main
# ============================================================
if __name__ == '__main__':
parser = argparse.ArgumentParser(
description='HCC-TACE DICOM → NIfTI 预处理',
formatter_class=argparse.RawDescriptionHelpFormatter,
epilog="""
示例:
# 检查 DICOM 数据
python preprocess_hcc_tace.py --inspect --dicom_root /path/to/hcc_tace_seg
# 完整转换
python preprocess_hcc_tace.py --dicom_root /path/to/hcc_tace_seg --output_root /path/to/output
"""
)
parser.add_argument('--dicom_root', type=str, default='.',
help='DICOM 数据根目录 (包含 HCC_XXX 子目录)')
parser.add_argument('--output_root', type=str, default='./processed_data',
help='输出目录')
parser.add_argument('--inspect', action='store_true',
help='检查模式:查看 DICOM 元数据而不转换')
parser.add_argument('--max_patients', type=int, default=10,
help='检查模式下显示的最大患者数')
parser.add_argument('--simple', action='store_true',
help='使用 pydicom+nibabel 简化转换(无需 dcm2niix)')
args = parser.parse_args()
if args.inspect:
inspect_mode(args.dicom_root, args.max_patients)
else:
process_dataset(args.dicom_root, args.output_root, args.simple)
然后运行检查:
BASH
# 安装依赖
pip install pydicom nibabel tqdm
# 检查模式 — 查看每个 series 的 Description,确认能否正确识别
python preprocess_hcc_tace.py --inspect --dicom_root ./hcc_tace_seg --max_patients 5
返回结果:
BASH
(mewm_py311) root@*****:/data/qklee/MeWM# python preprocess_hcc_tace.py --inspect --dicom_root ./hcc_tace_seg --max_patients 5
============================================================
患者: HCC_001
[PRE] Study: 00377
Series: 42120 | CT | arterial_ct | C-A-P (66 files)
Series: 76970 | CT | pre_ct | PRE LIVER (43 files)
Series: 99942 | SEG | tumor_mask | Segmentation (1 files)
[POST] Study: 49771
Series: 07012 | CT | pre_ct | Recon 2: PRE LIVER (87 files)
Series: 35194 | CT | other_ct | (92 files)
Series: 46705 | CT | other_ct | (92 files)
============================================================
患者: HCC_002
[PRE] Study: 84861
Series: 09935 | CT | pre_ct | PRE LIVER (24 files)
Series: 74386 | SEG | tumor_mask | Segmentation (1 files)
Series: 85789 | CT | arterial_ct | Recon 2: LIVER 3 PHASE (AP) (218 files)
[POST] Study: 93301
Series: 03644 | CT | pre_ct | PRE LIVER (27 files)
Series: 52402 | CT | arterial_ct | Recon 2: LIVER 3 PHASE (AP) (148 files)
============================================================
患者: HCC_003
[PRE] Study: 64595
Series: 18688 | CT | arterial_ct | Recon 2: LIVER 3 PHASE (AP) (190 files)
Series: 45632 | SEG | tumor_mask | Segmentation (1 files)
Series: 87624 | CT | pre_ct | PRE LIVER (20 files)
[POST] Study: 88989
Series: 76052 | CT | pre_ct | PRE LIVER (27 files)
Series: 94194 | CT | other_ct | Recon 2: 3 PHASE LIVER (ABD) (206 files)
============================================================
患者: HCC_004
[PRE] Study: 14785
Series: 39561 | SEG | tumor_mask | Segmentation (1 files)
Series: 40661 | CT | arterial_ct | Recon 3: LIVER 3 PHASE (AP) (79 files)
Series: 40949 | CT | pre_ct | PRE LIVER (20 files)
Series: 69759 | CT | arterial_ct | Recon 2: LIVER 3 PHASE (AP) (79 files)
[POST] Study: 54067
Series: 30056 | CT | pre_ct | PRE LIVER (20 files)
Series: 60035 | CT | other_ct | Recon 2: LIVER 3 PHASE A/P (170 files)
============================================================
患者: HCC_005
[PRE] Study: 20531
Series: 02396 | CT | other_ct | Recon 3: 3 PHASE LIVER (ABD) (81 files)
Series: 89106 | CT | other_ct | Recon 2: 3 PHASE LIVER (ABD) (81 files)
Series: 97639 | CT | other_ct | 3 PHASE LIVER (ABD) (81 files)
[POST] Study: 36548
Series: 06660 | SEG | tumor_mask | Segmentation (1 files)
Series: 81126 | CT | pre_ct | PRE LIVER (48 files)
Series: 85837 | CT | arterial_ct | LIVER 3 PHASE (AP) (89 files)
Series: 90548 | CT | arterial_ct | Recon 2: LIVER 3 PHASE (AP) (89 files)
然后开始跑转换:
PLAINTEXT
# 1. 先安装 dcm2niix(推荐,速度快)
conda install -c conda-forge dcm2niix
# 2. 执行转换
python preprocess_hcc_tace.py \
--dicom_root ./hcc_tace_seg \
--output_root ./HCC_TACE_processed
跑完后得到如下结果:
PLAINTEXT
============================================================
Step 3: 创建数据列表文件...
============================================================
成功配对: 102 个患者
写入 91 条记录 → ./HCC_TACE_processed/train_paired_all_hcc_under_50_new.txt
写入 11 条记录 → ./HCC_TACE_processed/val_paired_all_hcc_under_50_new.txt
============================================================
预处理完成!
============================================================
输出目录: ./HCC_TACE_processed
├─ HCC_tace_response_trans_pre/ (102 文件)
├─ HCC_tace_response_trans_pre_mask/ (102 文件)
├─ HCC_tace_response_trans_post/ (102 文件)
├─ HCC_tace_response_trans_post_mask/ (102 文件)
├─ train_paired_all_hcc_under_50_new.txt (91 条)
└─ val_paired_all_hcc_under_50_new.txt (11 条)
下一步:
1. 将输出目录 SCP/RSYNC 到服务器上的训练数据目录
2. 创建 cross_eval/liver/ 目录并放入数据列表文件
3. 确保 clinical data CSV 文件存在
4. 运行训练命令
后处理
接着处理下患者的信息,根据下载的HCC-TACE-Seg_clinical_data-V2.xlsx处理为csv:
PYTHON
#!/usr/bin/env python3
"""
将 HCC-TACE-Seg_clinical_data-V2.xlsx 转换为训练代码期望的 CSV 格式
用法:
python convert_clinical.py --xlsx HCC-TACE-Seg_clinical_data-V2.xlsx --output HCC_clinical_data.csv
"""
import argparse
import pandas as pd
import sys
def main():
parser = argparse.ArgumentParser(description='Excel → CSV 临床数据转换')
parser.add_argument('--xlsx', required=True, help='输入 Excel 文件')
parser.add_argument('--output', default='HCC_clinical_data.csv', help='输出 CSV 文件')
parser.add_argument('--inspect', action='store_true', help='仅查看 Excel 列名和示例数据')
args = parser.parse_args()
# 读取 Excel
xl = pd.ExcelFile(args.xlsx)
print(f"工作表: {xl.sheet_names}")
# 尝试读取每个工作表
for sheet in xl.sheet_names:
df = pd.read_excel(args.xlsx, sheet_name=sheet)
print(f"\n--- 工作表: {sheet} ---")
print(f"行数: {len(df)}, 列数: {len(df.columns)}")
print(f"列名: {list(df.columns)}")
print(f"\n前 3 行:\n{df.head(3).to_string()}")
if args.inspect:
return
# 自动选择第一个有效工作表
df = pd.read_excel(args.xlsx, sheet_name='data table')
# Excel 列 → 目标列 (仅文档参考)
# TCIA_ID→ID, Sex→sex, age→age, hepatitis→HBV, AFP→AFP,
# CPS→child, BCLC→BCLC, tumor_nodul→numoflesions, Tr_Size→diameter,
# Vascular invasion→VI, albumin/bilirubin=默认值
# albumin 和 bilirubin 无直接列,使用默认值
# chemotherapy 列可作为治疗文本描述
# ============================================================
# 构建输出 DataFrame
# ============================================================
out_df = pd.DataFrame()
out_df['ID'] = df['TCIA_ID'].astype(str).str.strip()
# 直接映射列
out_df['sex'] = pd.to_numeric(df['Sex'], errors='coerce') # 1=M, 0=F (或其他编码)
out_df['age'] = pd.to_numeric(df['age'], errors='coerce')
out_df['AFP'] = pd.to_numeric(df['AFP'], errors='coerce')
out_df['diameter'] = pd.to_numeric(df['Tr_Size'], errors='coerce')
# VI: Vascular invasion (0/1)
out_df['VI'] = pd.to_numeric(df['Vascular invasion'], errors='coerce')
# HBV: 从 hepatitis 列提取 (HBV only, HBV+HCV, etc.)
out_df['HBV'] = df['hepatitis'].astype(str).str.contains('HBV', case=False, na=False).astype(float)
# child: CPS 列 (Child-Pugh Score)
# 可能已是数值 (0=A, 1=B, 2=C) 或文本 (A, B, C)
cps_vals = df['CPS']
if cps_vals.dtype == object:
child_map = {'A': 0, 'B': 1, 'C': 2}
out_df['child'] = cps_vals.astype(str).str.upper().str.strip().map(child_map)
else:
out_df['child'] = pd.to_numeric(cps_vals, errors='coerce')
# BCLC: 可能已是数值 (0=A, 1=B, 2=C) 或文本 (Stage-A, Stage-B, A, B)
bclc_vals = df['BCLC']
if bclc_vals.dtype == object:
# 提取最后一级分类: Stage-A → A, Stage-B → B
bclc_clean = bclc_vals.astype(str).str.upper().str.strip().str.replace('STAGE-', '', regex=False)
bclc_map = {'A': 0, 'B': 1, 'C': 2, 'D': 3, '0': 0, '0.0': 0}
out_df['BCLC'] = bclc_clean.map(bclc_map)
else:
out_df['BCLC'] = pd.to_numeric(bclc_vals, errors='coerce')
# numoflesions: tumor_nodul 列 ("solitary"→1, "multinodular"→3)
nodule_map = {'solitary': 1, 'multinodular': 3, 'massive': 1, 'diffuse': 5}
out_df['numoflesions'] = df['tumor_nodul'].astype(str).str.lower().str.strip().map(nodule_map)
mask = out_df['numoflesions'].isna()
out_df.loc[mask, 'numoflesions'] = pd.to_numeric(df.loc[mask, 'tumor_nodul'], errors='coerce')
# albumin 和 bilirubin: Excel 中无对应列,使用默认正常值
out_df['albumin'] = 3.5 # g/dL
out_df['bilirubin'] = 0.8 # mg/dL
# ============================================================
# 数据清洗
# ============================================================
# 用中位数填充缺失值
for col in out_df.columns:
if col == 'ID':
continue
if out_df[col].dtype in ['float64', 'int64']:
out_df[col] = out_df[col].fillna(out_df[col].median())
# 任何剩余 NaN → 0
out_df = out_df.fillna(0)
# ============================================================
# 保存
# ============================================================
out_df.to_csv(args.output, index=False)
print(f"\n✓ 转换完成: {args.output}")
print(f" 行数: {len(out_df)}")
print(f" 列: {list(out_df.columns)}")
print(f"\n前 5 行预览:")
print(out_df.head(5).to_string())
if __name__ == '__main__':
main()
而后进行目录处理:
PYTHON
#!/usr/bin/env python3
"""
后处理修复脚本:
1. 将 HCC_TACE_processed 目录重组为 3 级路径(匹配训练代码期望格式)
2. 重新生成数据列表文件(含正确路径)
3. 检查临床数据 CSV 是否存在
4. 创建 cross_eval/liver/ 目录
"""
import os
import shutil
PROCESSED_DIR = './HCC_TACE_processed'
CROSS_EVAL_DIR = './Synthesis/Diffusion/cross_eval/liver'
CLINICAL_CSV_PATH = './HCC_clinical_data.csv'
# ============================================================
# Step 1: 重组目录为 3 级结构
# ============================================================
print("=" * 60)
print("Step 1: 重组目录为 3 级路径...")
print("=" * 60)
subdirs = [
'HCC_tace_response_trans_pre',
'HCC_tace_response_trans_pre_mask',
'HCC_tace_response_trans_post',
'HCC_tace_response_trans_post_mask',
]
# 扫描所有患者 ID
patient_ids = set()
for sd in subdirs:
sd_path = os.path.join(PROCESSED_DIR, sd)
if not os.path.isdir(sd_path):
continue
for f in os.listdir(sd_path):
if f.endswith('.nii.gz') and f.startswith('HCC_'):
# 提取 patient_id: HCC_XXX
# 文件名如: HCC_001_artery.nii.gz → HCC_001
# HCC_001_tumor.nii.gz → HCC_001
# HCC_001_ct.nii.gz → HCC_001
parts = f.split('_')
if len(parts) >= 2:
pid = f'{parts[0]}_{parts[1]}' # HCC_XXX
patient_ids.add(pid)
print(f"发现 {len(patient_ids)} 个患者")
for pid in sorted(patient_ids):
for sd in subdirs:
sd_path = os.path.join(PROCESSED_DIR, sd)
if not os.path.isdir(sd_path):
continue
# 创建患者子目录
pid_dir = os.path.join(sd_path, pid)
os.makedirs(pid_dir, exist_ok=True)
# 移动文件
for f in os.listdir(sd_path):
if not f.endswith('.nii.gz'):
continue
if f.startswith(pid): # HCC_001_artery.nii.gz
src = os.path.join(sd_path, f)
dst = os.path.join(pid_dir, f)
if os.path.isfile(src) and not os.path.lexists(dst):
shutil.move(src, dst)
print(" 目录重组完成 ✓")
# ============================================================
# Step 2: 重新生成数据列表文件(3 级路径)
# ============================================================
print("\n" + "=" * 60)
print("Step 2: 重新生成数据列表文件(3 级路径)...")
print("=" * 60)
# 从旧的数据列表文件读取配对信息
def read_old_data_list(filepath):
entries = []
if not os.path.exists(filepath):
return entries
with open(filepath) as f:
for line in f:
parts = line.strip().split('\t')
if len(parts) >= 6:
# 从旧路径中提取文件名
pre_ct_file = os.path.basename(parts[0]) # HCC_001_artery.nii.gz
pre_mask_file = os.path.basename(parts[1]) # HCC_001_tumor.nii.gz
post_ct_file = os.path.basename(parts[4])
post_mask_file = os.path.basename(parts[5])
# 提取 patient_id
pid = '_'.join(pre_ct_file.split('_')[:2]) # HCC_001
entries.append({
'patient_id': pid,
'pre_ct_file': pre_ct_file,
'pre_mask_file': pre_mask_file,
'post_ct_file': post_ct_file,
'post_mask_file': post_mask_file,
})
return entries
def write_data_list_3level(entries, output_path):
with open(output_path, 'w') as f:
for entry in entries:
pid = entry['patient_id']
# 3 级路径格式: DIR/patient_id/filename.nii.gz
pre_ct_rel = f'HCC_tace_response_trans_pre/{pid}/{entry["pre_ct_file"]}'
pre_mask_rel = f'HCC_tace_response_trans_pre_mask/{pid}/{entry["pre_mask_file"]}'
post_ct_rel = f'HCC_tace_response_trans_post/{pid}/{entry["post_ct_file"]}'
post_mask_rel = f'HCC_tace_response_trans_post_mask/{pid}/{entry["post_mask_file"]}'
treatment_text = 'TACE treatment'
line = '\t'.join([
pre_ct_rel, pre_mask_rel,
'', '',
post_ct_rel, post_mask_rel,
'', '', '',
treatment_text,
])
f.write(line + '\n')
print(f" 写入 {len(entries)} 条 → {output_path}")
old_train = os.path.join(PROCESSED_DIR, 'train_paired_all_hcc_under_50_new.txt')
old_val = os.path.join(PROCESSED_DIR, 'val_paired_all_hcc_under_50_new.txt')
train_entries = read_old_data_list(old_train)
val_entries = read_old_data_list(old_val)
print(f"训练集: {len(train_entries)} 条, 验证集: {len(val_entries)} 条")
# 覆盖写入(3 级路径)
write_data_list_3level(train_entries, old_train)
write_data_list_3level(val_entries, old_val)
# ============================================================
# Step 3: 检查真实临床数据 CSV
# ============================================================
print("\n" + "=" * 60)
print("Step 3: 检查临床数据 CSV...")
print("=" * 60)
if os.path.exists(CLINICAL_CSV_PATH):
import csv as _csv
with open(CLINICAL_CSV_PATH, 'r') as f:
reader = _csv.reader(f)
header = next(reader, [])
row_count = sum(1 for _ in reader) + 1 # +1 for header
print(f" ✓ {CLINICAL_CSV_PATH} 已存在 ({row_count} 行, 列: {header})")
else:
print(f" ⚠️ {CLINICAL_CSV_PATH} 未找到!")
print(f" 请先运行: python convert_clinical.py --xlsx HCC-TACE-Seg_clinical_data-V2.xlsx --output {CLINICAL_CSV_PATH}")
print(f" 或从本地上传真实临床数据 CSV 到此路径")
# ============================================================
# Step 4: 创建 cross_eval/liver/ 数据列表
# ============================================================
print("\n" + "=" * 60)
print("Step 4: 创建 cross_eval/liver/ 数据列表...")
print("=" * 60)
os.makedirs(CROSS_EVAL_DIR, exist_ok=True)
# 复制到 cross_eval/liver/
shutil.copy2(old_train, os.path.join(CROSS_EVAL_DIR, 'train_paired_all_hcc_under_50_new.txt'))
shutil.copy2(old_val, os.path.join(CROSS_EVAL_DIR, 'val_paired_all_hcc_under_50_new.txt'))
print(f" 数据列表已复制到 {CROSS_EVAL_DIR}/ ✓")
# ============================================================
# 完成总结
# ============================================================
print("\n" + "=" * 60)
print("后处理完成!")
print("=" * 60)
print(f"""
目录结构已修复:
{PROCESSED_DIR}/
├─ HCC_tace_response_trans_pre/{pid}/{pid}_artery.nii.gz
├─ HCC_tace_response_trans_pre_mask/{pid}/{pid}_tumor.nii.gz
├─ HCC_tace_response_trans_post/{pid}/{pid}_artery.nii.gz
├─ HCC_tace_response_trans_post_mask/{pid}/{pid}_tumor.nii.gz
├─ train_paired_all_hcc_under_50_new.txt
└─ val_paired_all_hcc_under_50_new.txt
临床数据: {CLINICAL_CSV_PATH}
交叉验证: {CROSS_EVAL_DIR}/
下一步 - 启动训练:
cd Synthesis/Diffusion
python3 train.py \\
dataset.name=liver_tumor \\
dataset.data_root_path=$(realpath ../{PROCESSED_DIR})/ \\
dataset.label_root_path=$(realpath ../{PROCESSED_DIR})/ \\
dataset.dataset_list=['liver'] \\
dataset.data_txt_path=cross_eval/ \\
dataset.uniform_sample=False \\
model.results_folder_postfix="liver" \\
model.vqgan_ckpt=pretrained_models/AutoencoderModel.ckpt
""")
生成模型训练
问题 1:Mask 编码不匹配
原始代码假设 | 实际数据 | |
|---|---|---|
类别数 | 3(背景/肝脏/肿瘤) | 2(背景/肿瘤) |
肿瘤值 | >=2 | 1 |
导致的连锁反应:
PYTHON
# ❌ 原始代码:直接把肿瘤(值=1)清零了
mask[mask == 1] = 0 # 所有肿瘤 voxel → 0!
mask[mask >= 2] = 1 # 永远找不到
# ✅ 修复
mask = (mask > 0).float() # 0=背景, 1=肿瘤
涉及位置:diffusion.py ×4、dataloader.py ×2
问题 2:CT 维度不一致
不同患者的 CT z 轴切片数 30~115 不等,同一患者的 pre/post CT 也可能不同。SpatialPadd 只能补不能裁。
PYTHON
# ✅ 新增 _match_spatial_shapes()
# 取所有 4 个图像(pre/post CT + mask)的最小空间维度
# 对较大者做 center-crop 对齐
问题 3:DICOM SeriesDescription 识别
TCIA 数据中 CT 相位描述不规范:
(AP)/C-A-P→ 动脉期PRE LIVER→ 平扫(跳过)3 PHASE LIVER (ABD)→ 多期混合Recon 2:前缀需递归解析
开始训练:
SHELLSCRIPT
conda activate mewm_py311
export HF_ENDPOINT=https://hf-mirror.com
cd /data/****/MeWM/Synthesis/Diffusion
python3 train.py \
dataset.name=liver_tumor \
dataset.data_root_path=/data/****/MeWM/HCC_TACE_processed/ \
dataset.label_root_path=/data/****/MeWM/HCC_TACE_processed/ \
dataset.dataset_list=['liver'] \
dataset.data_txt_path=cross_eval/ \
dataset.uniform_sample=False \
model.results_folder_postfix="liver" \
model.vqgan_ckpt=pretrained_models/AutoencoderModel.ckpt
训练中:
生存模型训练
首先处理数据:
PLAINTEXT
#!/usr/bin/env python3
"""
生成生存分析模型的数据列表文件
从已有的扩散模型数据列表 + 临床 CSV 中提取 survival_time 和 event_indicator
数据格式(Tab 分隔):
{pre_ct}\t{pre_mask}\t\t\t{post_ct}\t{post_mask}\t\t\t\t\t{survival_time}\t{event_indicator}
用法:
python prep_survival_data.py
"""
import os
import pandas as pd
import random
# 路径配置
EXCEL_PATH = 'HCC-TACE-Seg_clinical_data-V2.xlsx'
CLINICAL_CSV = 'HCC_clinical_data.csv'
DIFF_DATA_LIST = 'Synthesis/Diffusion/cross_eval/liver/train_paired_all_hcc_under_50_new.txt'
SURV_TRAIN_OUT = 'Survival/data/train.txt'
SURV_VAL_OUT = 'Survival/data/val.txt'
# ============================================================
# Step 1: 从 Excel 提取 OS 和 event 信息
# ============================================================
print("Step 1: 从临床 Excel 提取 OS + event...")
df = pd.read_excel(EXCEL_PATH, sheet_name='data table')
# 提取关键列
survival_info = {}
for _, row in df.iterrows():
pid = str(row['TCIA_ID']).strip()
# OS = 总生存期(天),转为月
os_days = float(row['OS']) if pd.notna(row['OS']) else 0.0
# event: 死亡=1, 存活/失访=0
# 优先用 Death 列,其次用 Censored 列的反义
if 'Death_1_StillAliveorLostToFU_0' in df.columns:
event = int(row['Death_1_StillAliveorLostToFU_0']) if pd.notna(row['Death_1_StillAliveorLostToFU_0']) else 0
elif 'Censored_0_progressed_1' in df.columns:
event = 1 - int(row['Censored_0_progressed_1']) if pd.notna(row['Censored_0_progressed_1']) else 0
else:
event = 0
survival_info[pid] = {
'survival_time': os_days,
'event_indicator': event,
}
print(f" 提取了 {len(survival_info)} 个患者的生存数据")
print(f" 示例: {list(survival_info.items())[:3]}")
# ============================================================
# Step 2: 从扩散模型数据列表读取配对,添加 survival 列
# ============================================================
print("\nStep 2: 生成生存分析数据列表...")
def create_survival_data(diff_list_path, output_path):
entries = []
with open(diff_list_path, 'r') as f:
for line in f:
parts = line.strip().split('\t')
if len(parts) < 6:
continue
# 提取 patient ID: 路径格式 HCC_tace_response_trans_pre/{pid}/{pid}_xxx.nii.gz
pre_ct_path = parts[0]
pid = pre_ct_path.split('/')[1] # {pid}
if pid not in survival_info:
print(f" [WARN] {pid} 没有生存数据,跳过")
continue
surv = survival_info[pid]
# 格式: pre_ct, pre_mask, (empty×2), post_ct, post_mask, (empty×4), survival_time, event
new_line = '\t'.join([
parts[0], # pre_ct
parts[1], # pre_mask
'', '', # unused
parts[4], # post_ct
parts[5], # post_mask
'', '', '', '', # unused
str(surv['survival_time']),
str(surv['event_indicator']),
])
entries.append(new_line)
os.makedirs(os.path.dirname(output_path), exist_ok=True)
with open(output_path, 'w') as f:
for e in entries:
f.write(e + '\n')
print(f" 写入 {len(entries)} 条 → {output_path}")
return len(entries)
os.makedirs('Survival/data', exist_ok=True)
n_train = create_survival_data(DIFF_DATA_LIST, SURV_TRAIN_OUT)
# 从训练集分出 10% 作为验证集
random.seed(42)
with open(SURV_TRAIN_OUT) as f:
lines = f.readlines()
random.shuffle(lines)
split = int(len(lines) * 0.9)
train_lines = lines[:split]
val_lines = lines[split:]
with open(SURV_TRAIN_OUT, 'w') as f:
f.writelines(train_lines)
with open(SURV_VAL_OUT, 'w') as f:
f.writelines(val_lines)
print(f"\n✓ 训练集: {len(train_lines)} 条")
print(f"✓ 验证集: {len(val_lines)} 条")
print(f"\n下一步训练(4 GPU):")
print(f" cd Survival")
print(f" python -W ignore train_ddp.py \\")
print(f" --dist-url tcp://127.0.0.1:$((RANDOM % 99999 + 10000)) \\")
print(f" --workers 4 \\")
print(f" --batch_size 4 \\")
print(f" --distributed \\")
print(f" --model_CT resnetMC3_18_wMask \\")
print(f" --aggregator TransMIL_seperate \\")
print(f" --lr 0.0001 \\")
print(f" --gpu 0,1,2,3 \\")
print(f" --survival_type OS \\")
print(f" --save_best \\")
print(f" --data_root /data/qklee/MeWM/HCC_TACE_processed/ \\")
print(f" --data_list_file ./data/train.txt \\")
print(f" --data_list_file_val ./data/val.txt")然后开始训练:
PLAINTEXT
cd /data/qklee/MeWM/Survival
python -W ignore train_ddp.py \
--dist-url tcp://127.0.0.1:$((RANDOM % 99999 + 10000)) \
--workers 4 --batch_size 4 --distributed \
--model_CT resnetMC3_18_wMask --aggregator TransMIL_seperate \
--lr 0.0001 --gpu 1 --survival_type OS --save_best \
--data_root /data/qklee/MeWM/HCC_TACE_processed/ \
--data_list_file ./data/train.txt --data_list_file_val ./data/val.txt目前看来复现效果很差,可能是数据集的原因,preprocess比较困难。
